
ALS (Amyotrophic Lateral Sclerosis) is a progressive neurodegenerative disease of the upper & lower motor neurons in the brainstem and motor cortex. In recent years, Mesenchymal stem cells (MSCs) and neural stem cells have been differentiated into specialized cells with specific functions needed for patients with ALS. MSCs and neural cells act as internal reservoirs in various tissues, crucial in the growth, development, survival, and repair and regeneration of lost function. Clinical trials and previous research in medical journals have shown that the typical life expectancy after diagnosis is 3 to 6 years. The actual cause of ALS and Motor Neuron Disease varies from patient to patient; however, it’s widely believed that the underlying issue for a majority of patients can be traced to the dysfunction or misregulation of a vital neuroprotein known as TDP-43.[1]
Motor neuron diseases (MND) are a family of conditions and include primary lateral sclerosis (PLS), amyotrophic lateral sclerosis (ALS), progressive muscular atrophy (PMA), monomelic amyotrophy (MMA), pseudobulbar palsy, Progressive Supranuclear Palsy (PSP), and progressive bulbar palsy.
Diagnosing ALS
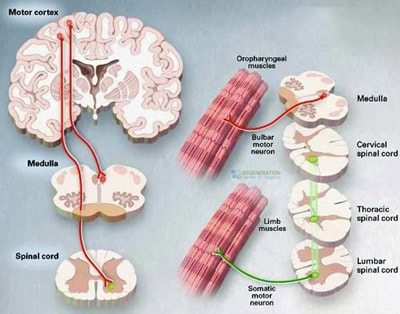
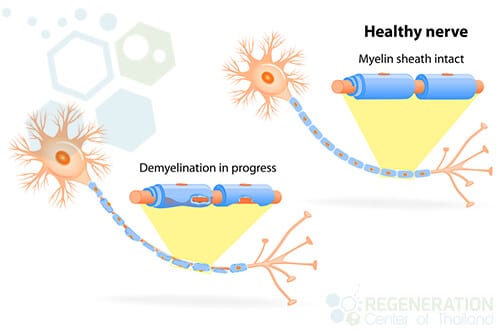 The diagnosis and classification of MND to ALS occur depending on how fast the patient’s disease progresses through stages, and if the condition is considered inherited/familial or sporadic/idiopathic. For most patients, symptoms of ALS are first experienced in the face or throat muscles. This early (onset) stage occurs when the lower motor neurons, nerve cells, and neurotransmitters in the brain stem (bulb/medulla oblongata) start to die. This early form of ALS is classified as “onset bulbar ALS.” Over time, the disease spreads to other areas, including the spinal cord motor neurons.
The diagnosis and classification of MND to ALS occur depending on how fast the patient’s disease progresses through stages, and if the condition is considered inherited/familial or sporadic/idiopathic. For most patients, symptoms of ALS are first experienced in the face or throat muscles. This early (onset) stage occurs when the lower motor neurons, nerve cells, and neurotransmitters in the brain stem (bulb/medulla oblongata) start to die. This early form of ALS is classified as “onset bulbar ALS.” Over time, the disease spreads to other areas, including the spinal cord motor neurons.
ALS vs. PLS and PMA
Classic symptoms of ALS occur in about 65% of all cases and affect both the upper and lower motor neurons. Classical ALS can be classified as bulbar-onset or spinal-onset (limb-onset) ALS disease. The classification of ALS is unique as the diagnosis of PLS (Primary lateral sclerosis) affects only the upper motor neurons in the arms and legs. In contrast, PMA (progressive muscular atrophy) only affects the lower motor neurons. Other variations of ALS include Respiratory-onset ALS, fail-leg syndrome (leg amyotrophic diplegia), flail-arm syndrome (brachial amyotrophic diplegia), and isolated bulbar ALS. Current clinical applications offer a better prognosis for Primary lateral sclerosis than classic ALS due to its slower rate of progress, which results in less functional decline and opens avenues for stem cell-based treatment. Males are more likely to develop young-onset ALS, while juvenile ALS is much more likely to be hereditary in nature than adult-onset ALS.
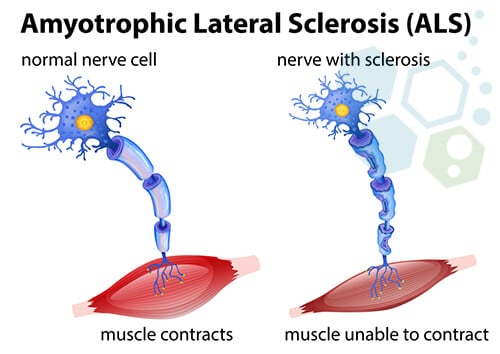
ALS, or Lou Gehrig’s disease, is a very aggressive neurological-degenerative disease that impacts the treatment of ALS and requires innovative approaches like stem cell-based therapy. nerve cells within the brain and the spinal cord. In particular, ALS refers to the degeneration of motor neurons that extend from the brain to the spinal cord and the muscles. ALS causes motor neuron degeneration, which may be mitigated by cell therapy. Cell signaling and neurotransmitters are often the first to die in a process called cell apoptosis. With it, the capability of the brain to instigate and manage voluntary muscle movements is similar to Parkinson’s Disease and MS. This will eventually result in total paralysis for the patient during the late stage of the illness.[2]
Signs and Symptoms of ALS
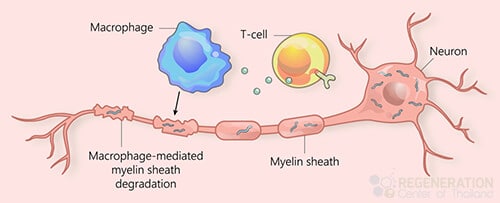
Due to the rapid degeneration of the upper and lower motor neurons, Neurodegenerative diseases such as ALS usually cause muscle atrophy, neuropathic pain, muscle spasticity, frequent muscle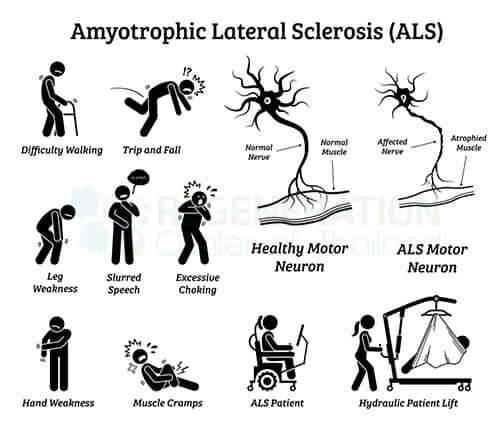 cramps, or spasms throughout the body. The symptoms are often mistaken for other conditions, such as peripheral neuropathy, but can also resemble those related to cell types affected in amyotrophic lateral sclerosis. Over time, patients with ALS lose the ability to control voluntary movements, bowel function, and respiratory failure, as well as the use of extraocular muscles that are responsible for the movement of the eyes. ALS isn’t considered to be. An example of an autoimmune disease that may be treated with cell-based therapy is multiple sclerosis, which shares some similarities with ALS. However, some stem cell researchers believe it may be partly due to a disorganized immune response and oxidative stress.
cramps, or spasms throughout the body. The symptoms are often mistaken for other conditions, such as peripheral neuropathy, but can also resemble those related to cell types affected in amyotrophic lateral sclerosis. Over time, patients with ALS lose the ability to control voluntary movements, bowel function, and respiratory failure, as well as the use of extraocular muscles that are responsible for the movement of the eyes. ALS isn’t considered to be. An example of an autoimmune disease that may be treated with cell-based therapy is multiple sclerosis, which shares some similarities with ALS. However, some stem cell researchers believe it may be partly due to a disorganized immune response and oxidative stress.
Along with physical degeneration, patients with ALS also suffer from behavioral or cognitive dysfunctions, which often get misdiagnosed as frontotemporal dementia, complicating the approach to therapies like mesenchymal stem cell transplantation. When cognitive or language changes are prominent, clinicians should also distinguish between neurodegenerative and vascular brain conditions because CSVD can affect memory, gait, and executive function without being a motor-neuron disease. Some common symptoms include repeated gestures or phrases, loss of inhibition, apathy, language problems, and verbal memory loss. About 50% of patients with bulbar-onset ALS experience depression and extreme emotional states where they laugh or cry for no apparent reason. Nociceptive pain is another common symptom, as patients report frequent muscle contractions in the cervical spine ( neck area), lower back pain, shoulder pain, or pressure in the stomach area.
Causes & Risk Factors of ALS
Despite rapid progress in understanding the causes, the cause of ALS (also known as Lou Gehrig’s disease) is relatively unknown but is generally attributed to hereditary (familial) or sporadic (environmental) causes. With advancements in molecular diagnostics, the genetic factors associated with ALS are better understood, making the treatment of ALS more effective than relying solely on ecological factors. There is no single specific cause of environmental ALS, but research has proven that the neurodegenerative damage associated with rare movement disorders, such as ALS, accumulates over long periods.
Chances of Getting ALS
Scientific studies have shown that familial (hereditary) ALS accounts for less than 10% of all confirmed cases. Inherited ALS is either an X-linked dominant pattern or an autosomal recessive pattern. Each parent carries one copy of the mutated gene but often shows no signs or symptoms. For this reason, autosomal recessive ALS is frequently misdiagnosed as environmental (sporadic) ALS. The most common markers for heredity ALS are C9orf72, SOD1, FUS, and TARDBP. Still, there are over 20 genes that are linked to familial ALS, and the only way to accurately identify genetic mutations is with DNA screenings.
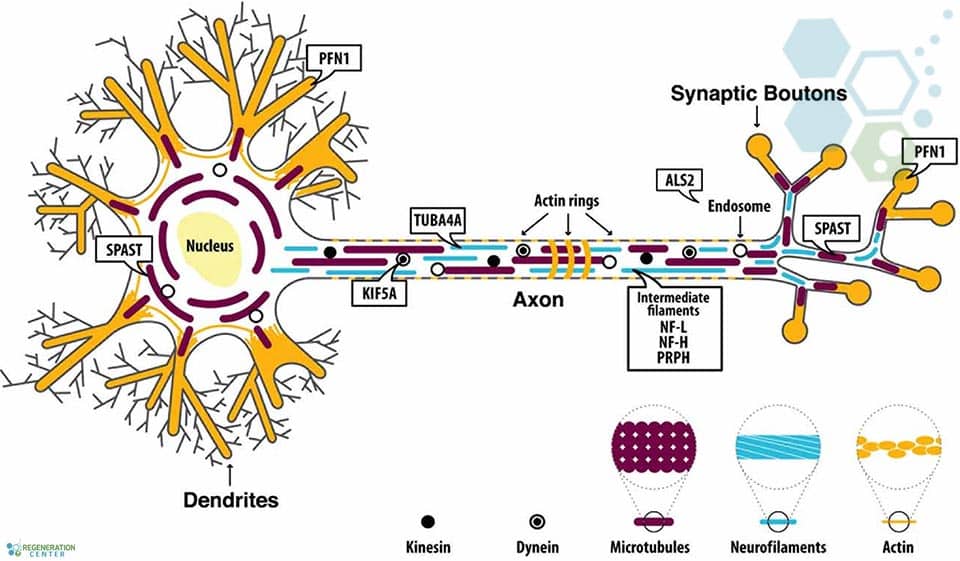
Genetic Testing for ALS
Since DNA testing was not prevalent in previous generations, it’s often difficult to predict unless proactively screened for. Clinical diagnosis for ALS typically requires mutations in at least 2 (or more) genes due to oligogenic inheritance. The regeneration center does not offer Gene therapies for ALS are being explored in various clinical studies. Still, it can provide insights into the efficacy of stem cell-based therapy for patients with amyotrophic lateral sclerosis. DNA sequencing tests are crucial for determining the best cell-based therapy options for patients with amyotrophic lateral sclerosis. For the 20 most common genetic causes of familial Amyotrophic Lateral Sclerosis, including ALS2, TBK1, SOD1, DCTN1, SPG11, TARDBP, KIF5A, FUS, OPTN, PFN1, SETX, CHCHD10, CHMP2B, MATR3, UBQLN2, VAPB, TFG, VCP, SIGMAR1, and SQSTM1.
Sporadic vs. Isolated ALS – ALS Research
Patients with sporadic ALS account for over 90% of all cases. It’s important to note that the symptoms of familial and sporadic ALS are nearly identical and can only be differentiated genetically. For patients with no known family history and mutated genes, environmental factors are typically the cause of the progression of ALS. ALS researchers have identified various factors, such as exposure to heavy metals and environmental toxins like lead, chemical exposure, pesticides, serum uric acid, DDT, organochlorine, dieldrin, and toxaphene, which are known foggy brain causes for damage to motor neurons, motor neuron death, and acute injuries to the central nervous system. Learn more about ALS-friendly diets.
Cause of ALS
Evidence is often inconclusive, but some potential environmental factors might include the following:
- Physical trauma due to an accident
- Repeat head injuries due to sports like boxing, MMA, NFL, and other contact sports can lead to long-term neurological issues similar to those seen in patients undergoing stem cell therapy in amyotrophic lateral sclerosis.
- Electromagnetic field exposure
- Severe traumatic brain injury is often misdiagnosed as chronic traumatic encephalopathy (CTE) or ataxia
ALS progression rates vary from person to person, but common symptoms include:
- Stiff finger joints
- Uneven fine motor skills
- Weakness in the hands due to muscle atrophy
- Problems with keeping the head up can be addressed through therapy for amyotrophic lateral sclerosis (ALS).
- Problems keeping good posture
- Weak muscles in the feet, ankles, or legs
- Benign fasciculation syndrome (BFS) is sometimes confused with symptoms seen in patients with amyotrophic lateral sclerosis.
- Sensory neuropathy can be a symptom that overlaps with conditions treated by cell-based therapies for ALS.
- Slurring of words during speech or trouble swallowing
- Aphasia and dysphasia
- Frequent muscle cramps
- Random twitching of the tongue, arms, or shoulders
- The trouble with the respiratory system and breathing normally
Stem Cell Treatment for ALS
The Regeneration Center treatment for ALS is a safe and effective functional medical treatment for complex neurodegenerative conditions such as MND & ALS, along with other therapies, are being developed in clinical trials and can be placed through the ALS Association. treatments for Brain Injuries, strokes, and Alzheimer’s all essentially focus on trying to slow disease progression, stopping the spread, and potentially reducing symptoms related to the illness. Neuro-stem cells and their possible role in the treatment of ALS are being researched extensively. Along with supportive glial cells, neural cell growth factors, and exosomes, these approaches can slow and reverse the consequences of ALS and restore the patient’s quality of life.[3]
TREATMENT RISKS & PRECAUTIONS
Please note that not all patients are suitable candidates for treating ALS with stem cells. Patients with advanced motor neuron damage, severe respiratory impairment, a history of cardiovascular disease, recent heart attacks, or other significant health issues might not be good candidates for treatment.Managing Disease Progression & Caring for ALS Patients
The Regeneration Center does not offer gene therapies, embryonic stem cells, induced pluripotent stem cells, or stem cells derived from animals, i.e., “Live Cells,” due to safety and efficacy concerns. Our enhanced neural stem cells and nerve growth factors are isolated and expanded to the required quantities and lineages, including glial-restricted progenitor cells (GRPs), oligodendrocyte progenitor cells (OPCs), astrocytes, and neurons.
Mesenchymal Stem Cells with Neural Growth Factors
After collection, the stem cells are concentrated and then isolated to measure the overall quantity and quality of the collected stem cells. Only certain types of activated neural progenitor stem cells can transform into the required cell types. These activated stem cells and neurotrophic factors can regenerate damaged tissues, such as the myelin sheath, which is needed in clinical therapies for ALS and in bone marrow treatments. spinal muscular atrophy.[4]
Management of ALS with Stem Cell Treatment
Number of Stages in Clinical Program: Multi-stage process using multi-combination infusions of glial restricted progenitor cells (GRP), oligodendrocyte neural progenitor cells (OPCs), Astrocytes exosomes are being investigated for their potential role in cell types relevant to ALS therapy, and neurons per treatment phase. The types of nerve cells and growth factors can include enhanced neural stem cells from differentiated cell lineage via IV Drip, Intrathecal, and lumbar puncture, per the patient’s needs. Learn about clinical trials for ALS and the efficacy of cell-based therapies being tested..

Benefits of Neural+ Stem Cells for ALS
Stem Cell Delivery Methods used: The Regeneration Center protocol combines multipotent stem cells from umbilical cord tissue (UC-MSC) and neural stem cells along with mesenchymal stromal cells, neurotrophic factors, and neural nerve cells that are delivered to the patient through IV Drips, Intramuscular injections, Intrathecal Injections (to bypass blood-brain barrier). In some cases, fluoroscopy-guided or Intraparenchymal injections may be required in a hospital setting for patients requiring direct injections into the spinal vertebra under local anesthesia. The patients are advised to remain in the recovery area for a few hours after the direct injection.
Physical Rehabilitation: Physical rehab is advised after therapy completion. Physical rehabilitation post-therapy can be provided upon request for 2-4 hours per day and up to 5 days per week. Medical visas and extended hotel or furnished-apartment accommodations for the patient and family can also be included upon request, especially for those seeking advanced therapies such as stem cell transplantation for amyotrophic lateral sclerosis.
Treatment of ALS with Motor Neurons
Treating Amyotrophic Lateral Sclerosis (ALS) with motor neurons derived from stem cells is an area of active research, offering hope for a condition that currently has no cure. ALS is a neurodegenerative disorder characterized by the progressive loss of motor neurons, the nerve cells responsible for controlling voluntary muscles. This loss leads to muscle weakness, paralysis, and eventually respiratory failure.
Stem Cell-Derived Motor Neurons
Motor neurons derived from stem cells, mainly induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs), are being explored as a potential treatment for ALS. The goal of these therapies is to replace the lost or damaged motor neurons in patients with ALS, potentially slowing or halting disease progression.
Critical approaches in ALS Treatment include:
- Direct Replacement: This approach involves differentiating stem cells into functional motor neurons in our stem cell laboratory and then transplanting them into the spinal cords of patients with ALS. The purpose of these transplanted neurons is to integrate into the patient’s nervous system and reactivate the functions of the lost neurons.
- Support of Existing Neurons: Stem cells or motor neurons might also be used to provide supportive factors that help to protect and maintain the surviving motor neurons in ALS patients. This approach requires transplanting cells and administering neurotrophic factors that support neuronal survival and function.
- Disease Modeling and Screening: Stem cells derived from ALS patients’ cells can be differentiated into motor neurons to create disease models; however, this remains in the experimental phase and is not yet available for clinical applications. These models are invaluable for studying ALS and screening potential biologicals that could slow disease progression or improve motor neuron survival.
Safety and efficacy for people with ALS
Several essential factors come into play when considering the safety and efficacy of using motor neurons derived from stem cells to treat ALS. Safety concerns primarily revolve around the risk of immune rejection, where the body might attack the transplanted cells if they are not HLA-matched to the patient’s cells. There is also a risk of tumor formation, especially with pluripotent stem cells such as iPSCs and ESCs, as these cells can proliferate rapidly. Efficacy concerns focus on whether the transplanted motor neurons can survive, integrate into the patient’s central nervous system, and function properly, especially in stem cell therapy for amyotrophic lateral sclerosis. This includes ensuring they connect correctly with muscles and other neurons, which is crucial for helping restore lost functions. Stem cell research is ongoing to determine whether these treatments can safely and effectively slow or halt the progression of ALS, with early results showing promise but also highlighting the need for more research to overcome these challenges.
Treatment for ALS Guidelines for 2026
Please note, we do not offer gene therapies for ALS. For approved patients, the treatment will require at least 2 weeks for cell transplantation. Due to the significant differences in the extent of damage and the candidate’s stage of recovery, our medical team will need to review the potential candidate’s medical records and eligibility for treatment and travel to Thailand. Upon completion of the medical review, a complete clinical plan will be provided that will include the specifics of the therapy, including the total number of nights required and the total medical-related costs and stem cell transplant fees (excluding accommodations or flights). To begin the qualification process for the multi-stage ALS stem cell transplantation, please prepare your recent medical records, such as a list of current medications (radical/radical, edaravone, riluzole), all functional rating scales, Brain MRIs, CT Scans, or PET Scans (Preferred), and contact us today.
Published Clinical Citations
[1] ^ Kwon, Min-Soo, Min-Young Noh, Ki-Wook Oh, Kyung-Ah Cho, Byung-Yong Kang, Kyung-Suk Kim, Young-Seo Kim, and Seung H Kim are contributing to ALS research. 2014. The immunomodulatory effects of human mesenchymal stem cells on peripheral blood mononuclear cells in ALS patients. Journal of Neurochemistry, no. 2 (July 31) is a crucial date for the phase 2 clinical studies on ALS therapies. doi:10.1111/jnc.12814. https://www.ncbi.nlm.nih.gov/pubmed/24995608
[2] ^ Mazzini, Letizia, Angelo Vescovi, Roberto Cantello, Maurizio Gelati, and Alessandro Vercelli. 2016. Stem cell therapy for ALS. Expert opinion on biological therapy, specifically regarding stem cell treatment in amyotrophic lateral sclerosis, is no. 2 (January 9). doi:10.1517/14712598.2016.1116516. https://www.ncbi.nlm.nih.gov/pubmed/26558293
[3] ^ Poungvarin, N, and A Viriyavejakul. 1991 marked significant advancements in the understanding of stem cell-based therapy for neurological diseases, leading to the exploration of cell transplantation in amyotrophic lateral sclerosis. Motor neuron disease in Thailand: the clinical aspects of 77 patients. Journal of the Medical Association of Thailand = Chotmaihet thangphaet, no. 4. https://www.ncbi.nlm.nih.gov/pubmed/1940701
[4] ^ Thomsen, Gretchen M, Genevieve Gowing, Soshana Svendsen, and Clive N Svendsen. 2014. The past, present, and future of stem cell clinical trials for ALS. Experimental neurology (March 6). doi:10.1016/j.expneurol.2014.02.021. https://www.ncbi.nlm.nih.gov/pubmed/24613827