

Pulmonary fibrosis is the thickening and scarring of the lung tissue. The term idiopathic denotes an unknown cause and spontaneous onset. Though it is not entirely understood what causes idiopathic pulmonary fibrosis and diffuse parenchymal lung failure, the disease process is well-explained in various medical literature and case histories.[1]
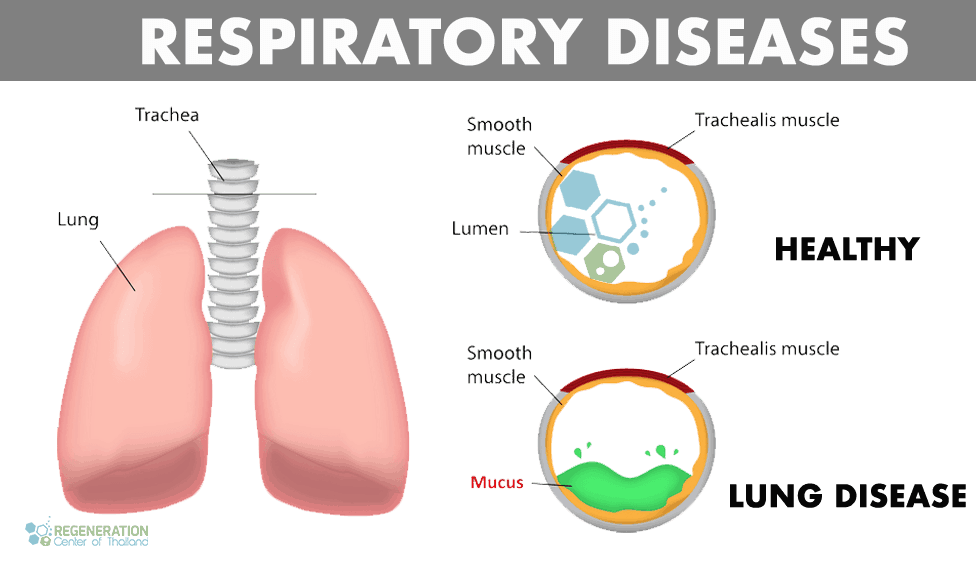
Healthy vs. Diseased Lungs
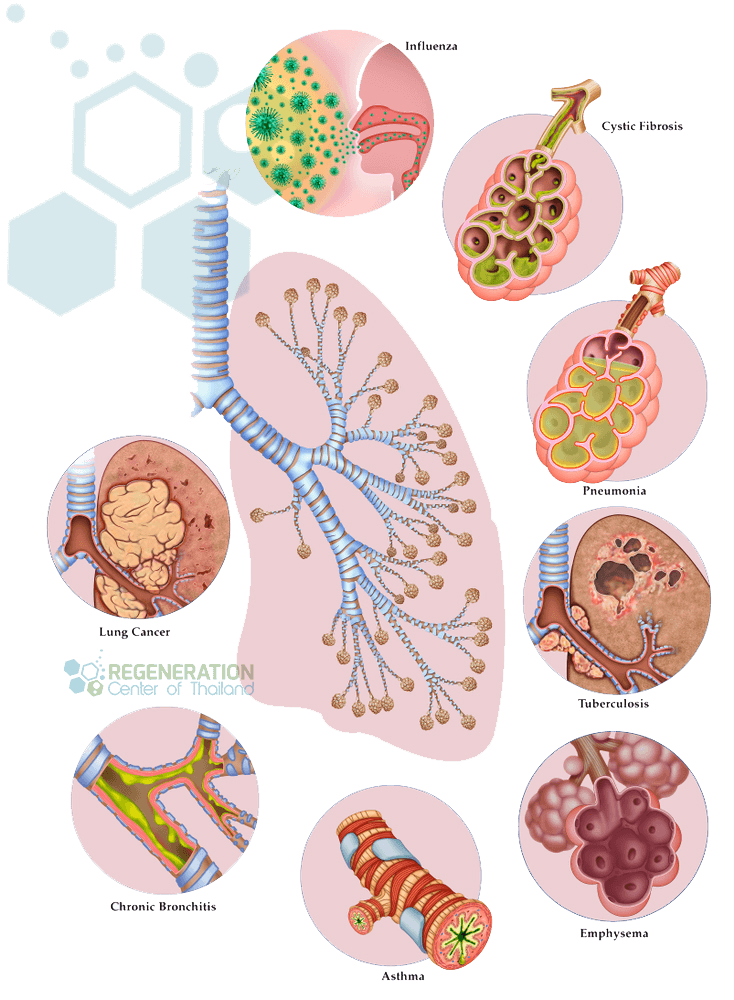
Interstitial Pulmonary Fibrosis
Diffuse parenchymal lung disease (DPLD) & Interstitial lung disease (ILD) are groups of lung diseases that affect the tissue and spaces around the air sacs (alveoli) in the lungs (interstitium). This family of lung disorders arises from lung injury that fails to heal properly for various reasons. Areas of the lungs that are prone to injuries include:
- Pulmonary capillary endothelium
- Basement membrane – a specialized form of extracellular matrix (ECM)
- Alveolar epithelium – functional & physical barrier of dangerous environmental agents
- Perilymphatic lung tissue
- Perivascular tissue
Managing Alveolar Fibrosis – Fibroid Lung Disease
Under normal circumstances, the human body can initiate self-repair of the lungs using paracrine cell signaling. However, this routine lung repair process malfunctions in some patients with interstitial lung disease, leading to thickening of the tissue around alveolar air sacs and scarring. Thickened scarred lung tissue makes it difficult for oxygen to pass back into the bloodstream, damaging the entire cardiovascular system and lungs. Pulmonologists use the term Interstitial lung disease to distinguish these lung diseases from others, such as COPD and obstructive airway diseases. If left untreated, interstitial lung disease results in fibrotic scarring of the lungs, known as pulmonary fibrosis. Idiopathic pulmonary fibrosis usually gets diagnosed after radiographic lung scans show pleural-based fibrosis with honeycombing in the lungs and proliferating fibroblasts (fibroblastic foci)
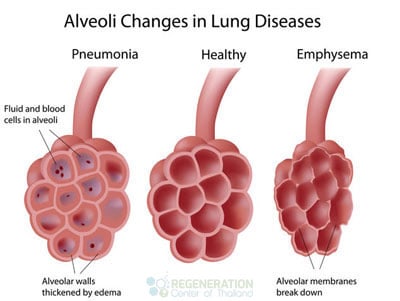
IPF and fibroid lung disease usually start with the presence of inflammation in the alveoli. The alveoli are the tiny air sacs in the lungs where carbon dioxide and oxygen exchange occurs. The fragile lining of the alveoli allows this gas exchange. Once these alveoli become inflamed and damaged, the body’s immune response is activated to heal them.[2]
This causes scarring and thickening of the alveolar walls. Unfortunately, the thickened and scarred alveoli cannot perform efficiently during gas exchange, transporting less oxygen into the body and expelling less carbon dioxide.
Signs & Symptoms of Pulmonary Fibrosis
Some of the common signs and symptoms that are present in patients diagnosed with idiopathic pulmonary fibrosis are:
- Severe shortness of breath is caused by heart disease or decreased oxygen supply due to inefficient affected alveoli. It gets worse with physical activity as the body tries to get more oxygen.
- Constant Dry cough.
- Clubbing sensations of the fingers and toenails. Clubbing is the swelling in the finger’s base and is usually accompanied by cyanosis (bluish discoloration of the nails). This is a sign of hypoxia or decreased oxygen level in the tissue. This sign often manifests later in the disease progression and end-stage pulmonary fibrosis.
- Aching muscles and joints
- Poor appetite and unexplained weight loss
- Reduction of lung capacity or airflow obstructions resulting in premature fatigue
- Frequent Chest Pain
- Constant tiredness and lingering fatigue
Causes of Lung Scarring – ILD & IPF
Since the underlying cause of Idiopathic lung fibrosis is often unknown, patients usually wonder how they get fibrotic scarring of the lungs. For most patients, the most common predisposing factors of developing interstitial pulmonary disease & alveolar fibrosis are:
- Cigarette smoking is a common cause of interstitial pulmonary fibrosis
- Genetic – Hereditary lung disease, including fibrotic scarring of the lungs
- Exposure to various inorganic toxins, chemicals, and pollutants such as asbestos, beryllium (berylliosis), crystalline silica dust (silicosis)
- Exposure to organic compounds, dust, fungus, or mold (hypersensitivity pneumonitis)
- Reaction to drugs or medications such as antibiotics, chemotherapy drugs & cardiac dysrhythmia medications (antiarrhythmic)
- Viral infections such as pneumocystis pneumonia (PCP), tuberculosis, atypical pneumonia, chlamydia trachomatis & syncytial virus
- GERD (Gastroesophageal reflux disease). This is when the stomach contents of a patient go back to the esophagus. There is a risk of aspiration of these stomach contents into the trachea, which can, in turn, lead to a lung infection.[3]
- Connective Tissue Disorders & autoimmune response due to conditions such as Lupus, rheumatoid arthritis, polymyositis, dermatomyositis, lymphangitic carcinomatosis, antisynthetase syndrome (inflammation), and systemic sclerosis
- Inflammatory bowel diseases such as ulcerative colitis and Crohn’s disease
- Undifferentiated spondyloarthritis
- Idiopathic (non-identifiable), i.e., acute interstitial pneumonia (Hamman-Rich syndrome), Idiopathic pulmonary fibrosis & sarcoidosis
Diagnosing Pulmonary Fibrosis
Diagnosing interstitial lung diseases and pulmonary fibrosis requires diagnostic tests and physical exams. A pulmonologist uses several diagnostic tools to understand the signs & better
Symptoms of patients with interstitial lung disease. Depending on the stage and severity of a patient’s condition, lung specialists typically order the following tests:
- Pulmonary function tests for measuring decreased diffusion capacity (DLCO) to see if any restrictions exist
- Lung radiology scans use X-ray or High-Resolution Chest CT (HRCT) to detect patterns of opacities in lung tissue caused by thickened, scarred lung tissue.
- An arterial blood test can assess the lungs’ ability to deliver oxygen to the body’s tissues.
- Lung biopsy involves taking a sample of lung tissue and studying it to identify scarring, thickening, or other signs of malignancy. Biopsies can include a transbronchial biopsy or the more invasive surgical lung biopsy.
- Genetic testing for pulmonary fibrosis and Cystic Fibrosis – May prevent the need for surgical lung biopsy
Screening for Mutations that may cause Familial Pulmonary Fibrosis
The Lung Regeneration Center offers a comprehensive list of genetic screening tests to detect mutations in genes that lead to familial lung diseases. Tests for the following genes are currently available to screen for congenital autosomal recessive & autosomal dominant interstitial lung diseases:
ABCA3, FLCN, SFTPB, SFTPC,CSFR2A, CSFR2B, COPA,TSC1, TTF1, TSC2, DOCK8, STAT3, FoxF1, PGM3, SPINK5, METRS, MRS, MTRNS, CMT2U, ILFS2, ILLD, mENA, NISBD2, RTEL1, PIG61, ERBB, ERBB1, HER1, SPG70, TERC, TERT & PARN
Genetic screening tests are recommended for family members of patients who have already been diagnosed. There are currently no available gene therapies to treat autosomal interstitial lung diseases, but several clinical trials are underway with the hopes of one day curing genetic lung diseases. To help reduce any risk of developing fibroid lung disease, ILD, or Pulmonary fibrosis, consider including pulmonary tests as part of your annual checkup, refraining from smoking, and getting regular exercise to keep the lungs and heart as healthy as possible.
Traditional vs. Modern Treatments for ILD & Lung Fibrosis
In the past, there were traditionally minimal options for treating chronic progressive lung diseases such as COPD and idiopathic pulmonary fibrosis. Even today, several clinical trials are underway to treat interstitial lung diseases, familial pulmonary fibrosis, and idiopathic lung sclerosis using modified gene therapies. Still, as of today, no effective pharmaceutical medication-based cures are approved for treatment. In some cases, steroids are prescribed to help decrease inflammation or mucosal secretions (Acetylcysteine) to reduce symptoms. Still, the effects are temporary and do nothing to the underlying cause of the disease. Lung transplants are another option for some, but they are considered very invasive and high-risk and are usually only offered to advanced end-stage pulmonary fibrosis patients. Although it has its limitations, modified MSC+ lung stem cells provide a natural alternative to lung transplants for pulmonary fibrosis and anti-fibrotic medications.[4]
3 Years After Treatment for Lung Fibrosis with Stem Cells
Stop the Hardening of Lung Disease & Alveolar Fibrosis Naturally
Your Stem cells are essentially the body’s pharmacy. Stem cells are the building blocks of your entire body. These potent cells are called “totipotent,” meaning they can differentiate into any cell, tissue, or bone in the human body. When appropriately used, UC-MSC+ Stem Cells can help address loss of lung capacity/reduced lung function by targeting the damaged or limited alveolar lining at the root of the disease. [5] For various reasons (Cigarette smoking, Viral infections, genetics), the body’s natural ability to regenerate is being blocked, causing the types of complications that manifest themselves as fibrosis and lung hardening disease.
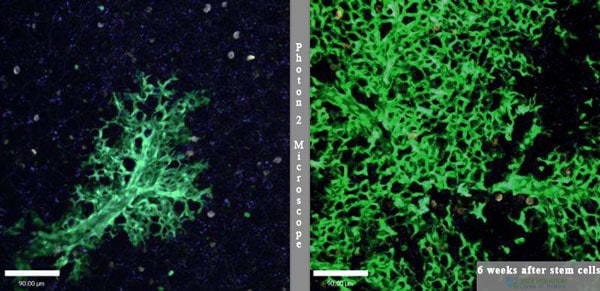
The Regen Center treatment for IPF lung sclerosis uses isolated & enhanced lung stem cells that aim to re-promote the natural healing of previously damaged alveolar cells, reduce fibrotic scarring in the interstitium lining the lungs, and remove lung scarring from pneumonia. The goal of the enhanced UC-MSC+ stem cell treatment for pulmonary fibrosis and lung hardening disease is to help the body bypass fibrotic scarring by regenerating healthy tissues and blood vessels through angiogenesis. The latest lung regeneration treatment offers a safe, natural alternative to lung transplants by regenerating lung tissue affected by fibrotic scarring and honeycombing through increased myofibroblast levels.
Multi-Stage Lung Cell Delivery for Respiratory Diseases
Interstitial lung diseases & progressive pulmonary fibrosis pose a significant challenge for pulmonologists and lung bioengineering. Diseases such as asthma, popcorn lungs, “Bronchiolitis obliterans,” Idiopathic pulmonary fibrosis, lung cancer treatment, COPD, idiopathic interstitial pneumonia, Cystic fibrosis, emphysema & hyaline membrane disease are just some of the diseases that can be managed effectively via lung stem cell therapies. Due to the complexity and locations of the diseased regions, it can be challenging to effectively deliver cell therapeutic agents to the needed areas of the diseased lungs. The Lung Regeneration Center offers stem cells for lung disease patients using a combination of UC-MSC+ cells to halt the rapid decline in lung capacity. Our unique cell delivery methods target diseases using multiple vectors, including guided radiography (when necessary), Intravenous, direct injection, or intrathecal routes for painless and effective delivery of endogenous lung progenitor cells and lung epithelial stem cells to both the airway and vascular systems of the lungs.
TREATMENT RISKS & PRECAUTIONS
Please note that not all patients are suitable candidates for treating idiopathic pulmonary fibrosis (IPF), lung fibrosis, or bronchiectasis with stem cells. Patients with advanced lung disease, severe respiratory impairment, significant comorbidities, multi-organ failure, or other critical health conditions might not be good candidates for treatment.Stem Cell Treatment for Bronchiectasis
Bronchiectasis and bronchial fibrosis are often the leading causes of respiratory morbidity and mortality. In medical diagnosis, bronchiectasis severity is based on the extent of damage to the patient’s airways. Some patients have cylindrical/ tubular fibrosis, while others have cystic or varicose fibrosis. Tubular and Cylindrical bronchiectasis are usually the most common and most treated forms of bronchiectasis. In contrast, cases of cystic bronchiectasis are more challenging and sometimes untreatable because the irreversible destruction of lung bronchi and alveoli makes them difficult to treat. Stem cell treatment for bronchiectasis and bronchial fibrosis using a combination protocol of isolated mesenchymal stem cells (UC-MSC+), bronchiolar epithelium, and airway basal cells.

Guidelines for Treating Chronic Pulmonary Fibrosis
The total number of UC-MSC+ Stem Cell infusions will depend on the severity of restrictive lung disease, and multiple stages may be necessary for patients with interstitial lung scarring or a diffuse parenchymal diagnosis. For optimal results, multiple cell-delivery methods will be required. Due to the progressively degenerative nature of restrictive lung diseases, most patients will require enhanced cell transplants derived from immune-compatible cells. For patients with multiple simultaneous issues, extensive honeycombing, or severe respiratory failure, multiple treatments may be necessary. The isolated and expanded UC-MSC+ lung cells are pre-screened for infectious diseases, sterility, and potency. For patients with severe respiratory failure, aggressive combination treatment may be required.
Lung Regeneration Requirements & Lung Rehabilitation
Post-Treatment Lung Rehabilitation: Pulmonary rehabilitation services are optional but are highly recommended after any treatment. We can offer a complete integrative rehabilitation program for local patients in partnership with local hospitals in Bangkok. Pulmonary rehabilitation is also beneficial for patients experiencing severe difficulty breathing or severe lung hardening. Smoking is also much discouraged for patients with this condition, and active smokers will not qualify for our treatment. The rehab packages are available to all candidates as needed (2-5 hours per day, six days per week) . Medical treatment visas and hotel accommodations for long-term stays can also be provided upon request.
Due to varying degrees of respiratory disease, our pulmonary specialists must better understand patients’ needs to qualify potential candidates. Our initial review can be completed online, and the medical assessment takes about 7 business days. Suitable candidates looking to reverse lung scarring naturally using Stem Cells will require a minimum of 2 weeks in Thailand. To begin the qualification process, please prepare your recent pulmonary tests, such as spirometry exams, lung function tests, and radiology scans from MRI or (HRCT) High-Resolution chest CT scans, and contact us today.
Published Clinical Citations
[1] ^ Pongrakhananon, Varisa, Sudjit Luanpitpong, Todd A Stueckle, Liying Wang, Ubonthip Nimmannit, and Yon Rojanasakul. 2014. Carbon nanotubes induce apoptosis resistance of human lung epithelial cells through FLICE-inhibitory protein. Toxicological sciences : an official journal of Thailand Society of Toxicology, no. 2 (November 19). doi:10.1093/toxsci/kfu251. https://www.ncbi.nlm.nih.gov/pubmed/25412619
[2] ^ Liu, Ming, Dunqiang Ren, Dong Wu, Jian Zheng, and Wenwei Tu. 2015. Stem Cell and Idiopathic Pulmonary Fibrosis: Mechanisms and Treatment. Current stem cell research & therapy, no. 6. https://www.ncbi.nlm.nih.gov/pubmed/25986617
[3] ^ Ghadiri, Maliheh, Paul M Young, and Daniela Traini. 2015. Cell-based therapies for the treatment of idiopathic pulmonary fibrosis (IPF) disease. Expert opinion on biological therapy, no. 3 (December 15). doi:10.1517/14712598.2016.1124085. https://www.ncbi.nlm.nih.gov/pubmed/26593230
[4] ^ Chambers, Daniel C, Debra Enever, Nina Ilic, Lisa Sparks, Kylie Whitelaw, John Ayres, Stephanie T Yerkovich, Dalia Khalil, Kerry M Atkinson, and Peter M A Hopkins. 2014. A phase 1b study of placenta-derived mesenchymal stromal cells in patients with idiopathic pulmonary fibrosis. Respirology (Carlton, Vic.), no. 7 (July 9). doi:10.1111/resp.12343. https://www.ncbi.nlm.nih.gov/pubmed/25039426
[5] ^ Barczyk, Marek, Matthias Schmidt, and Sabrina Mattoli. 2015. Stem Cell-Based Therapy in Idiopathic Pulmonary Fibrosis. Stem cell reviews and reports, no. 4. doi:10.1007/s12015-015-9587-7. https://www.ncbi.nlm.nih.gov/pubmed/25896401