

Cystic fibrosis, or CF, is a genetic disease that affects the lungs and digestive system by causing them to become clogged with thick, sticky mucus. For a child to be born with CF, both parents must be carriers of the defective gene that causes it. When both parents carry the faulty gene, each child they have has a 25% chance of having the condition, a 25% chance of being healthy, and a 50% chance of being a carrier. Cystic fibrosis, which used to claim its victims in infancy or early childhood, has now become a killer of people in their 30s because treatments of the infections that characterize the disease have improved. But despite these advances, there has been slow progress in treating the underlying mutations that affect the vast majority of patients, which is a defect in a single gene that interferes with the fluid balance in the surface layers of the airways and leads to a thickening of mucus, difficulty breathing, and repeated infections and hospitalizations.[1] Learn more about gene testing for CF. 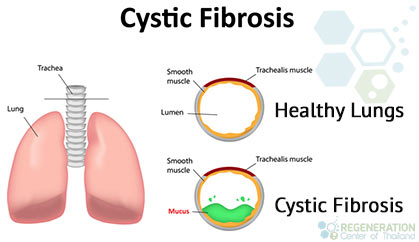
What is the Cause of Cystic Fibrosis?
CF is caused by mutations in the CFTR (cystic fibrosis transmembrane conductanceo have premature stop regulator) protein [2]. In one mutation class, the CFTR is prematurely terminated because stop codons, signals to end protein production, occur earlier than expected. Mutations in CFTR proteins (W1282X and G542X) cause the mRNA (messenger RNA) to have premature stop codons. This means that the CFTR protein is not produced in its full form because a stop codon halts translation before the protein is fully synthesized [3]. Therefore, people with cystic fibrosis who have these mutations have a non-functional CFTR.
The CFTR protein must be complete for it to be functional. To target this issue, the stop codon must not appear prematurely in the production sequence. If this can be corrected by gene editing, it could offer a potential form of gene therapy for people with cystic fibrosis.
Symptoms of Cystic Fibrosis
Some common symptoms of CF can include:
- Wheezing
- Shortness of breath
- Inability to exercise
- Persistent, mucus-laden coughing
- Intestinal blockage and constipation
- Frequent lung infections
- Stuffy nose
- Poor weight gain
In recent years, awareness of cystic fibrosis has grown dramatically. Since the mid-20th century, when research began revealing its mechanisms and several famous people were found to suffer from the disease, it has taken center stage as a focus of research and scientific inquiry. Today, diagnosing cystic fibrosis in adults is categorized as delayed-onset cystic fibrosis, which is also called cystic fibrosis in the diagnosis of babies [4].
Traditional Treatment Options for CF
Due to the disease’s genetic nature, there is no cure for cystic fibrosis, and current treatments are symptomatic.
Currently, cystic fibrosis and chronic autosomal recessive diseases are treated with the following:
- Antibiotics to prevent and treat chest infections
- Medications to widen the airways
- Medicines to thin mucus within the lungs
- Medical devices to clear mucus from the lungs
- Medicines to reduce inflammation
- Medications to help patients absorb food, diet modifications, and supplements
A lung transplant may even become necessary for patients with severe cystic fibrosis.
Gene Therapy for Cystic Fibrosis
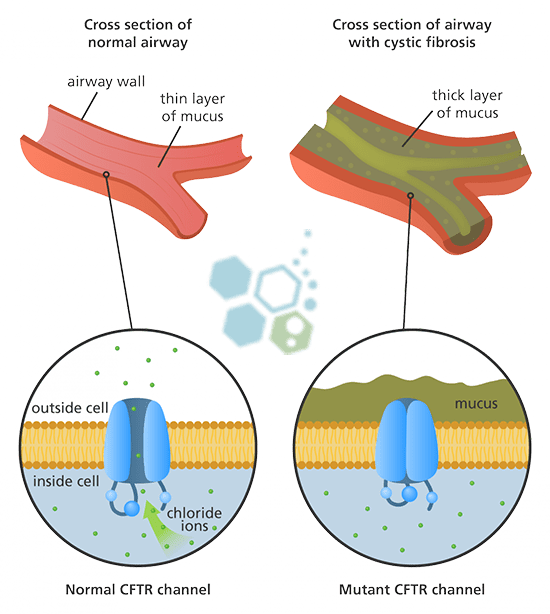 The cystic fibrosis CFTR gene ( transmembrane conductance regulator) contains all the instructions for making the CFTR protein. When the instructions are mutated, the production of this vital CFTR protein is affected. In patients with cystic fibrosis, mutations in the CFTR gene can result in insufficient CFTR protein or incorrect CFTR protein. Each defect in the production of these proteins leads to a cascading set of issues that can affect the lungs and other organs.
The cystic fibrosis CFTR gene ( transmembrane conductance regulator) contains all the instructions for making the CFTR protein. When the instructions are mutated, the production of this vital CFTR protein is affected. In patients with cystic fibrosis, mutations in the CFTR gene can result in insufficient CFTR protein or incorrect CFTR protein. Each defect in the production of these proteins leads to a cascading set of issues that can affect the lungs and other organs.
Since the initial discovery of the CFTR gene in 1990, researchers have been seeking new ways to correct the mutations in the gene that cause cystic fibrosis. Although progress was slower than anticipated, scientific breakthroughs have accelerated advances in gene therapies, gene transfer, and gene replacement over the past decade.
Gene therapy works by introducing a corrected version of the CFTR gene back into the person’s body. Although the previously mutated copies of the CFTR gene would still be present, the corrected DNA copies would allow cells to make normal CFTR proteins, resulting in recovery.
Stem Cell Treatment for Cystic Fibrosis
Stem cell therapy offers new hope to patients seeking a viable treatment for cystic fibrosis. Mesenchymal stem cells are the body’s “master cells,” capable of becoming any specialized cell the body needs. Specifically, multipotent cells in the body can differentiate into a wide range of cell types, including bronchial, bronchiolar, and alveolar epithelial cells. Multipotent cells can be sourced from the patient or cord tissue or cord blood and are often used to treat lung airway diseases such as COPD, bronchiectasis, and Pulmonary fibrosis. While pluripotent stem cells and iPSCs can become any cell in the body, they are challenging to manage and can lead to uncontrolled division, which can result in cancer.
New research suggests that cystic fibrosis treatment could be available sooner than we think. Studies show that umbilical cord tissue may have the potential to treat the disease and restore healthy lung function. For now, the research explores using stem cells to help counteract the dysfunction and overproduction of mucus in damaged bodily systems [5].
Stem cells can help patients with cystic fibrosis in a few ways, including:
- Stem cells can replace the affected cells with healthy ones.
- Multipotent bronchioalveolar stem cells isolated from lung tissue may provide another promising approach for successful stem cell therapy.
- Stem cells can help repair and regenerate in mitotic cellular compartments.
- Bronchial stem cells, which can be used for maintaining bronchial, bronchiolar, and alveolar epithelia
- Stem cells can home to the damaged respiratory epithelium and undergo regeneration.
- Stem cell therapy can colonize the airways, proliferate there, and differentiate into columnar cells that cover a sufficient portion of the airway epithelium.
Struggling with Cystic Fibrosis Symptoms? Here’s How to Breathe Easier
Living with cystic fibrosis can feel like an uphill battle, with persistent symptoms like difficulty breathing, chronic infections, and overwhelming fatigue taking a toll on daily life. For many, the thick mucus clogging the airways makes breathing labored, creating a breeding ground for recurring lung infections, leading to hospital visits and disruptions to life’s rhythm. Fatigue from the constant fight against these symptoms can leave even the simplest tasks exhausting. But breakthroughs in treatment options are offering renewed hope. Stem cell therapy is a promising approach that targets the root cause of cystic fibrosis rather than just managing the symptoms. By regenerating damaged lung tissue and reducing inflammation, stem cells can help restore healthy lung function. In addition to stem cell lifestyle adjustments, tailored pulmonary rehabilitation programs, proper nutrition, and personalized medication plans can support easier breathing and improved energy levels. For those struggling with these symptoms, these integrative solutions can be life-changing, offering relief and the possibility of a healthier, more active future.
Non-Surgical Lung Scarring Treatments for Cystic Fibrosis
Lung scarring, or pulmonary fibrosis, is a serious complication for many cystic fibrosis patients, significantly impairing lung function and quality of life. Traditional treatments often focus on symptom management but do little to reverse the underlying damage. Innovative approaches like stem cell therapy are changing this landscape by addressing the scarring at its core. Mesenchymal stem cells (UC-MSCs) possess powerful regenerative properties that target inflammation and promote the repair of damaged lung tissue. These cells can reduce fibrosis, restore elasticity to lung structures, and support the regrowth of healthy alveolar cells, which are necessary for effective oxygen exchange. By reducing scar tissue and enhancing lung function, stem cell therapy offers new hope to patients with limited options. When combined with advanced pulmonary rehabilitation programs, these treatments improve breathing capacity and help patients regain the ability to engage in everyday activities without constant respiratory distress. This cutting-edge solution is paving the way for better outcomes and a brighter future for those living with cystic fibrosis.
2026 Guidelines for Cell-Based Cystic Fibrosis Therapies
The total number of UC-MSC+ Stem Cell infusions will depend on the severity of airway damage, and multiple stages may be necessary for those with excessive interstitial scarring, chronic inflammation in the lung & diffuse parenchymal disease. Due to the degenerative nature of CF, most patients will also require endogenous airway progenitor cells in combination with mesenchymal stromal cells.
Pulmonary Rehabilitation Post Therapy
In Thailand, Pulmonary rehabilitation services are optional but highly recommended after any treatment. For patients living in Thailand, we can offer a comprehensive integrative rehabilitation program in partnership with local hospitals. Pulmonary rehabilitation can also be beneficial for patients with severe difficulty breathing or severe lung hardening. The post treatment rehab packages are available for all candidates 2-5 hours per day and up to six days per week . Medical treatment visas and hotel accommodations for long-term stays can also be provided upon request.
TREATMENT RISKS & PRECAUTIONS
Not all patients are suitable candidates for treating cystic fibrosis & lung diseases with stem cells. Patients with advanced lung disease, severe scarring/respiratory impairment, significant comorbidities, multi-organ failure, or other critical health conditions might not be good candidates for treatment. The Regeneration Center does not offer gene therapies for Cystic fibrosis.Loss of CFTR function can affect multiple organs, including the pancreas, lungs, liver, kidney, and intestines. Due to varying degrees of severity and stages, our pulmonary specialists will need to understand the patient’s current medical needs to qualify any potential candidates for treatment. Our initial review can be done online or in person. Suitable candidates seeking treatment for lung scarring with stem cells will require at least 2 weeks to complete the protocol. To begin the qualification process, please prepare your recent pulmonary tests, such as spirometry exams, lung function tests, and radiology scans from MRI or (HRCT) High-Resolution chest CT scans, and contact us today.
Published Clinical Citations
[1] ^Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013 Dec 5;13(6):653-8. doi: 10.1016/j.stem.2013.11.002. PMID: 24315439.
[2] ^ Conese M, Beccia E, Castellani S, Di Gioia S, Colombo C, Angiolillo A, Carbone A. The long and winding road: stem cells for cystic fibrosis. Expert Opin Biol Ther. 2018 Mar;18(3):281-292. doi: 10.1080/14712598.2018.1413087. Epub 2017 Dec 8. PMID: 29216777.
[3] ^Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BL. Lung organoids: current uses and future promise. Development. 2017 Mar 15;144(6):986-997. doi: 10.1242/dev.140103. PMID: 28292845; PMCID: PMC5358104.
[4] ^ Conese M, Rejman J. Stem cells and cystic fibrosis. J Cyst Fibros. 2006 Aug;5(3):141-3. doi: 10.1016/j.jcf.2006.02.001. Epub 2006 Mar 6. PMID: 16574502.
[5] ^Weiss DJ. Stem cells and cell therapies for cystic fibrosis and other lung diseases. Pulm Pharmacol Ther. 2008 Aug;21(4):588-94. doi: 10.1016/j.pupt.2007.11.004. Epub 2007 Dec 7. PMID: 18203640; PMCID: PMC4201362.